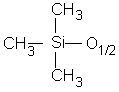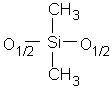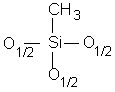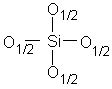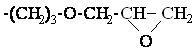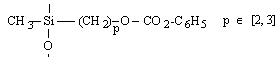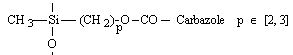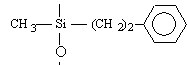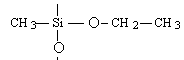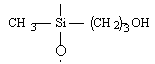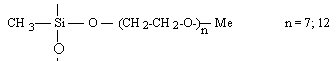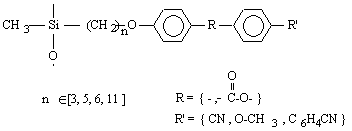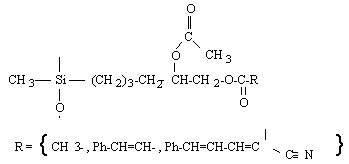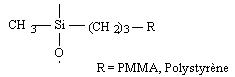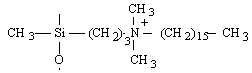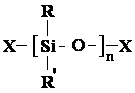
Formation chimie des polymères :
Synthèse des silicones ou polysiloxanes
(Texte extrait de la thèse de P. CANCOUËT avec l'accord de l'auteur)
Ce sont le français Friedel et l'américain Crafts qui synthétisèrent les premiers polyalkylsiloxanes entre 1865 et 1872 (124,125), mais l'industrie des silicones est née pendant la deuxième guerre mondiale (12,125) . Aujourd'hui, les silicones connaissent un grand développement grâce à leurs propriétés physico-chimiques remarquables qui sont dues essentiellement au squelette inorganique constitué par l'enchaînement Si-O-Si. Celui-ci offre une très bonne stabilité thermique et une excellente inertie chimique dues à la remarquable solidité de la liaison Si-O. Cette liaison est en particulier très résistante à l'oxydation et à l'hydrolyse (la liaison Si-O présente une énergie de liaison de 88,2 kcal.mol-1 contre 83,1 kcal.mol-1 pour la liaison C-C (126,127) ). D'autre part, leur combustibilité reste très limitée, la variation de leur viscosité sous l'effet de la température est faible, ainsi que leur tension superficielle (20 dynes.cm-1 ), leur résistance aux cisaillements intenses et prolongés est excellente et leur perméabilité aux gaz est forte (12,125) . Enfin, leur absence de toxicité et leur inertie chimique permettent l'emploi de nombreux types de silicones en cosmétologie, dentisterie, médecine, chirurgie, pharmacie et dans les industries alimentaires (13,125,128,129) . Ces polysiloxanes sont constitués par l'enchaînement de motifs :
dans lesquels les substituants R et R' peuvent être H, CH3 , C2H5 , C6H5 , CH2=CH-, etc... et X un substituant ou un groupement chimique de nature variable.
Pour simplifier l'écriture des dérivés siloxaniques, une convention généralement admise consiste à désigner leurs motifs par l'un des quatre symboles M, D, T ou Q (12,125) (Tableau II-1).
Ainsi, le tétramère cyclique (octaméthylcyclotétrasiloxane) est désigné par D4 et un polydiméthylsiloxane de n motifs D terminé à chaque extrémité par un motif triméthylsilyle sera symbolisé par MDnM.
Si les polysiloxanes sont des polymères originaux en raison des nombreuses propriétés que nous venons d'énumérer, il est souvent intéressant de les fonctionnaliser avec divers groupements organofonctionnels pour obtenir de nouvelles propriétés ayant des applications dans des domaines divers. Dans la littérature, nous trouvons de nombreux exemples de synthèses ou applications mettant en jeu des polysiloxanes fonctionnalisés qui peuvent être différenciés :
Par la position dans la chaîne macromoléculaire des fonctions réactives.
Par la nature chimique des fonctions portées par ces polysiloxanes.
|
SYMBOLE |
MOTIF |
UNITE |
|
M
|
Monofonctionnel |
|
|
D
|
Difonctionnel |
|
|
T
|
Trifonctionnel |
|
|
Q
|
Tétrafonctionnel |
|
Tableau II-1 : Symboles utilisés pour identifier les silicones.
Nous pouvons donc distinguer deux grandes catégories de polysiloxanes fonctionnalisés :
o Polysiloxanes a,w -difonctionnalisés servant à la préparation de copolymères séquencés ou "à blocs" ou de résines tridimensionnelles. Dans ce cas, les fonctions réactives sont portées par les extrémités de la chaîne. Elles sont susceptibles de donner lieu à des couplages avec d'autres types de fonctions portées par des polymères de nature différente (par condensation, addition, ou substitution) pour donner des copolymères séquencés (Tableau II-2).
|
Nature des extrémités des PDMS a,w - difonctionnels |
Copolymères obtenus à partir des PDMS a,w - difonctionnels
|
|
PDMS a, w diaminopropyl
|
poly(DMS-b-uréthane).......................... poly(DMS-b-imide)................................ poly(DMS-b-aramide)........................... poly(DMS-b-arylester)........................... poly(DMS-b-esteraramide).................. |
|
PDMS a, w dihydroxyalkyl m Î [1,5] |
poly(DMS-b-uréthane)..........................
poly(DMS-b- e -caprolactone)................
|
|
PDMS a, w diépoxyalkyl
|
poly(DMS-b-ester insaturé)..................
poly(DMS-b-sulfone).............................
|
|
PDMS a, w dihydrogéno
-H
|
poly(DMS-b-(tétraméthyl-p-silphénylènes))....................................... poly(DMS-b-styrène et a- méthyl-styrène)........................................ poly(DMS-b-oxyéthylène).................... poly(DMS-b-isobutylène)..................... poly(DMS-b-ester insaturé).................. poly(DMS-b-sulfone)............................. |
|
PDMS a, w -dichloro -Cl |
poly(DMS-b-ester insaturé)................. poly(DMS-b-carbonate)........................ |
|
PDMS a, w dicarboxyalkyl |
poly(DMS-b-carbonate)........................
poly(DMS-b-aramide)........................... |
|
PDMS a, w dibischloroformiate -R'-O-CO-Cl |
poly(DMS-b-ester)................................. |
Tableau II-2 : Polydiméthylsiloxanes a,w -difonctionnalisés servant à la préparation de copolymères à blocs.
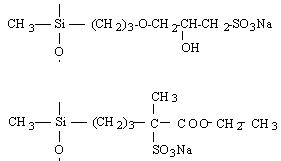
o Polysiloxanes à fonctions latérales : Ils constituent la seconde classe de polyorganosiloxanes fonctionnalisés. Ces polymères se caractérisent par la fixation sur la chaîne principale de groupes chimiques qui peuvent être de nature très variable (Tableau II-3). Les propriétés physico-chimiques d'un polymère fonctionnalisé sont les propriétés relatives au polyorganosiloxane auxquelles s'ajoutent les propriétés particulières des groupes pendants. Les polysiloxanes à fonctions sulfonate que nous avons préparés se placent dans cette seconde catégorie de polymères fonctionnalisés à groupes pendants. Les fonctions étant greffées sur des polysiloxanes à motifs SiH répartis le long de la chaîne macromoléculaire et appelés [poly(oxydiméthylsilylène - co - oxy-hydrogénométhylsilylène)] en nomenclature systématique. Le greffage des fonctions s'obtient par la réaction d'hydrosilylation des oléfines très utilisée dans l'industrie et au laboratoire.
|
Nature de la fonction latérale
|
|
Carbonate |
|
Carbamate |
|
Phényléthyl |
|
Ethoxy |
|
Alcool |
|
Polyéther |
Tableau II-3 : Polysiloxanes à fonctions latérales.
|
Nature de la fonction latérale
|
|
Groupements mésogènes |
|
Dérivés halogénés et esters |
|
Alkylsulfonate |
|
Groupements photoréticulables |
|
Polymères |
|
Ammonium quaternaire |
Tableau II-3 : Polysiloxanes à fonctions latérales (suite).
Il s'agit de polymères dont la formule générale est la suivante :
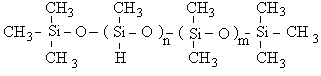
Pour simplifier l'écriture, nous désignerons par D les motifs diméthylsiloxane -(CH3)2Si-O-, par DH les motifs hydrogénométhylsiloxane -(CH3)HSi-O- et par M les bouts de chaînes (CH3)3Si-O-. Le copolymère ci-dessus sera donc noté : MDHnDm M.
Les méthodes de synthèse des polysiloxanes sont essentiellement au nombre de trois :
l'hydrolyse des chlorosilanes ;
la polymérisation anionique par ouverture de cycles ;
la polymérisation cationique par ouverture de cycles.
Nous passerons rapidement sur les deux premières, car seule la polymérisation cationique peut s'appliquer à la synthèse de notre copolymère.
Cette méthode consiste à effectuer l'hydrolyse ménagée du dichlorodiméthylsilane (164) . Les fonctions chlorosilane sont transformées en fonctions silanol avec formation d'acide chlorhydrique in situ . Ces fonctions silanol instables en présence d'acide chlorhydrique vont ensuite s'autocondenser pour donner des oligomères linéaires et cycliques (165,166) . Une telle synthèse présente de nombreux inconvénients dont le plus grand est l'importance des réactions parasites : la forte concentration d'acide chlorhydrique au sein du milieu réactionnel provoque de multiples réactions d'attaque des liaisons Si-O-Si des chaînes en croissance. Les liaisons Si-C peuvent également être attaquées, ce qui provoque des pontages intermoléculaires et l'apparition de chaînes ramifiées. De plus, cette réaction est inutilisable dans le cas de copolymères comportant des motifs hydrogénométhylsiloxane, car la présence d'eau et d'acide entraînerait des réactions secondaires de branchement avec la fonction silane (167,168) (équations II-1 et II-2). La réaction II-1 est connue depuis longtemps (167) , car elle se manifeste par la formation d'hydrogène. C'est pourquoi les hydrogénosilanes doivent toujours être manipulés dans des solvants secs.
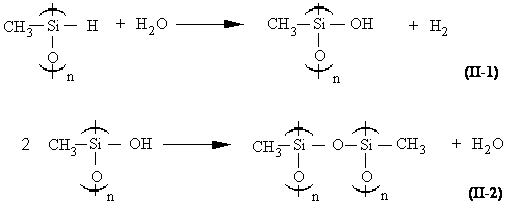
La polymérisation anionique des cyclosiloxanes est connue depuis longtemps (169-174) . Toutefois, de la même façon que pour l'hydrolyse des chlorosilanes, la polymérisation anionique par ouverture de cycles doit être écartée dans le cas de copolymères comportant des motifs hydrogénométhylsiloxane. En effet, d'après Lee (174) , la très grande instabilité de ce type de fonction en milieu basique entraîne la formation de réseaux tridimensionnels. Le mécanisme serait un transfert d'hydrure lors de l'attaque du centre actif silanolate sur un motif -Si(CH3)H-O- du monomère ou de la chaîne de polymère (équation II-3)

La polymérisation des cyclosiloxanes peut être amorcée soit par des acides de Brönsted (minéraux ou organiques), soit par des acides de Lewis.
En ce qui concerne les polymérisations amorcées par les acides de Lewis, nous pouvons citer comme amorceurs FeCl3 (175) , SnCl4 (176) , SbCl3 (177) , AlCl3 , BF3 (127) .
La réaction est possible avec D3 et D4 , mais à température élevée (de l'ordre de 200 °C ). Les réactions en masse sont très lentes. L'obtention de polymère nécessite l'intervention d'un cocatalyseur : HCl, H2O, C2H5OH (127) .
De nombreux acides minéraux (nitrique, phosphorique et perchlorique (178) ) amorcent la polymérisation des cyclosiloxanes. L'acide sulfurique (166,177,178) est l'un des plus employés, mais il doit être utilisé en solution concentrée, ce qui conduit à des problèmes de solubilité dans les siloxanes. On peut aussi utiliser un catalyseur "terre acide", c'est-à-dire un support minéral finement divisé et traité avec de l'acide chlorhydrique ou de l'acide sulfurique (140) .
Des acides organiques comme l'acide trifluoroacétique (179) , l'acide p-toluène-sulfonique (180) et l'acide trifluorométhanesulfonique (triflique) (181-186) sont aussi utilisés. L'acide triflique présente l'avantage d'être efficace à faible concentration (10-4 à 10-3 mol.l-1 ) et de donner des solutions homogènes.
La première étude approfondie de la polymérisation cationique d'un cyclosiloxane a été réalisée avec D3 en présence de CF3SO3H dans le dichlorométhane par Chojnowski et al. (181-183,186) .
Plus récemment, Lebrun et al. (183-185) ont proposé un mécanisme de la polymérisation de D4 amorcée par CF3SO3H dans CH2Cl2 (éventuellement en présence de M2 ). Ce mécanisme peut être transposé aux synthèses que nous avons réalisées.
L'étude réalisée par Lebrun (184) montre que la polymérisation de D4 amorcée par CF3SO3H conduit à la formation de quatre familles de siloxanes : au début de la réaction deux familles de cycles (D5 -D9 , D10 -D70 ) se forment parallèlement à un bas polymère de masse moléculaire d'environ 20.000 g.mol-1 . Puis, un haut polymère (M~100.000 g.mol-1 ) apparaît après une période d'induction, tandis que la quantité de cycles diminue jusqu'à atteindre son équilibre.
Pour interpréter ces résultats, deux types de réactions ont été proposés :
Une réaction de polymérisation par ouverture de cycles conduisant à la formation du bas polymère et aux produits cycliques par réaction de "rétroscission" ("back-biting").
Une réaction de polycondensation donnant le haut polymère.
a ) Réaction de polymérisation en chaîne
Amorçage . La première étape de la réaction est l'acidolyse de la liaison siloxane (équation II-4), immédiatement suivie par l'homocondensation des extrémités silanol catalysée par l'acide (équation II-5). La formation du centre actif résulte de la protonation d'un groupement ester triflique par une molécule d'acide (équation II-6). Cet ester ainsi activé serait le siège d'une attaque nucléophile par un oxygène d'un oligomère siloxanique (D4 étant le plus abondant au début) pour conduire à un ion oxonium tertiaire, centre actif de la polymérisation (équation II-7).
Acidolyse :
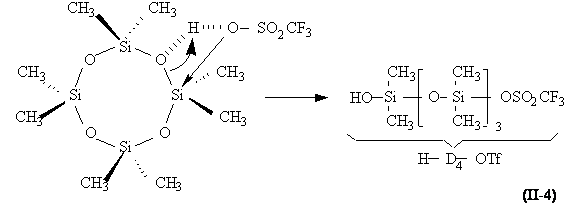
Homocondensation des extrémités silanol :
2 TfO-D4-H + H + , TfO - , x H2O TfO-D8 -Tf + H + , TfO - , (x+1) H2O
(II-5)
Formation des centres actifs :
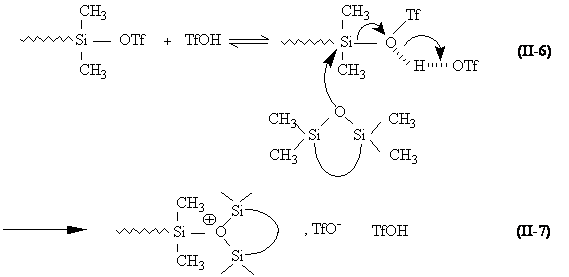
Schéma II-1 : Amorçage de la polymérisation de D4 par CF3SO3H (TfOH).
Propagation. L'allongement de la chaîne s'effectue par addition du monomère sur le centre actif oxonium (équation II-8).
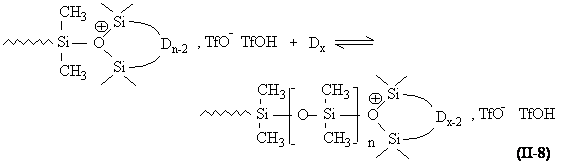
Cependant, des réactions secondaires intramoléculaires interviennent de façon importante. En effet, les atomes d'oxygène de la chaîne en formation ont une nucléophilie peu différente de celle des oligomères cycliques. Il se produit donc des réactions de dépolymérisation intramoléculaire ou de "rétroscission" qui mènent à la formation (dès le début de la réaction) de cycles de toutes tailles (équation II-9). Chacun de ces cycles pouvant à son tour réagir avec un centre actif, on aboutit à un équilibre entre les macromolécules linéaires et chaque type de cycle.
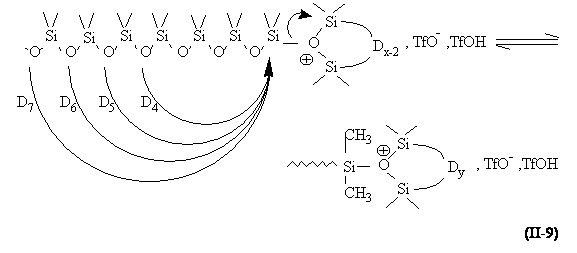
Chaque espèce cyclique possède une constante de cyclisation K x dont la valeur décroît avec le nombre x de motif unitaire (187-188) . Wright et Semlyen (189) ont donné les pourcentages massiques des espèces cycliques à l'équilibre thermodynamique (en masse) pour chacun des homopolymères MDHnM (polyhydrogénométhylsiloxane) et MDmM (Tableau II-4).
|
Unité monomère |
Pourcentage massique de cycles (Dx ) contenus dans un haut polymère à l'équilibre thermodynamique. |
|||
|
|
x = 3-5 |
x = 6-18 |
x = 19- 8 |
Total |
|
DH |
4,5 |
3,4 |
4,6 |
12,5 |
|
D |
10,0 |
3,6 |
4,7 |
18,3 |
Tableau II-4 : Pourcentages massiques de cycles pour le polyhydrogénométhylsiloxane (DH ) et le polydiméthylsiloxane (D) à l'équilibre thermodynamique (à 383 K) (189) .
Terminaison. La désactivation du centre actif se fait par recombinaison de l'anion triflate avec l'ion oxonium et mène à la formation de macromolécules a , w -diester (équation II-10).
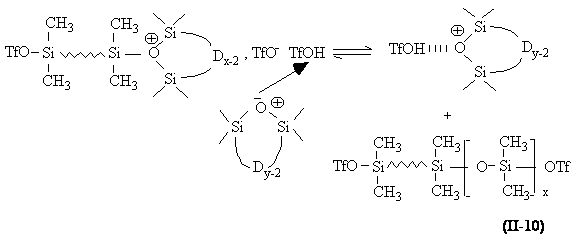
b ) Réaction de polycondensation
Cette réaction débute par une hydrolyse (équation II-11) suivie par une hétérocondensation entre les extrémités ester et les extrémités silanol, qui conduit à la formation de chaînes de haut polymère. Cette réaction peut être intramoléculaire et conduire à la formation de cycles (équation II-12) ou intermoléculaire (équation II-13).
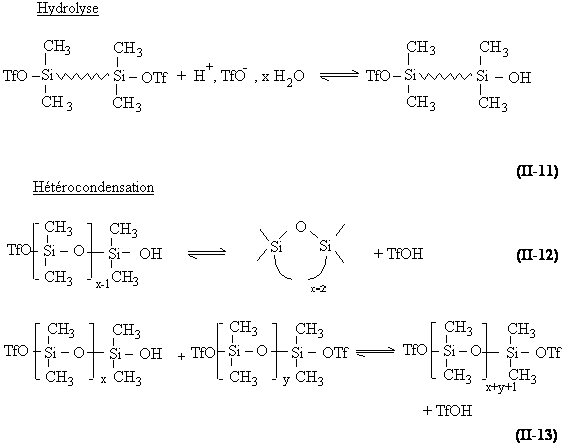
Afin de contrôler la masse molaire du polymère, il est nécessaire d'adjoindre au milieu réactionnel un agent télogène ou "limiteur de chaîne" tel que l'hexaméthyldisiloxane (HMDS ou M2 ).
Le mode d'action de M2 peut être simplement interprété par une réaction d'addition sur le centre actif, en compétition avec l'étape normale de propagation (équation II-14). La basicité de M 2 étant supérieure à celle de D4 , cette réaction est favorisée. De plus, il est logique que M 2 subisse aussi une acidolyse au début de la réaction, parallèlement à celle de D4 .
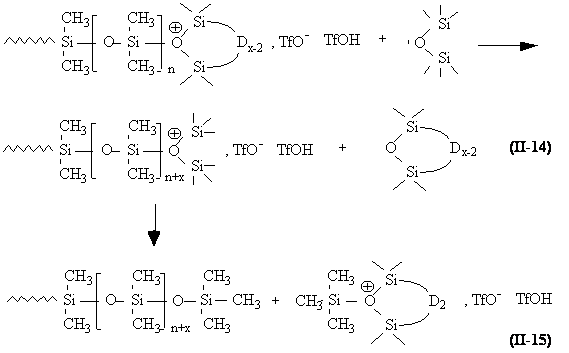
Ainsi, à l'équilibre, toutes les macromolécules linéaires possèdent des extrémités triméthylsilyle. Le DPn du polymère qui sera finalement récupéré après élimination d'une fraction x de cycles volatils sera donc donné par :
DPn = 4 [D4] (1-x)/ [HMDS]
(II-16)
La polymérisation cationique de D4 en présence de HMDS permet donc la synthèse de PDMS de masse molaire moyenne en nombre contrôlée. Un autre intérêt de cette méthode est que l'utilisation de disiloxanes, analogues au HMDS et présentant une fonction sur chaque atome de silicium, permet la synthèse de PDMS a,w-difonctionnels.
Les références bibliographiques pourront être fourni en envoyant un e-mail à : contact@atomer.fr